Preamble
Introduction
The SpatialData
package provides an R interface to the SpatialData framework, a
unified ecosystem for handling spatial omics data. Developed as part of
the scverse project (Virshup et al. 2023), SpatialData
aims to solve the challenges of integrating diverse spatial
datasets—including imaging, spatial transcriptomics, and proteomics—by
employing the OME-NGFF (Next
Generation File Format) standard (Marconato
et al. 2025).
The Python implementation and core specifications can be found at the official SpatialData website.
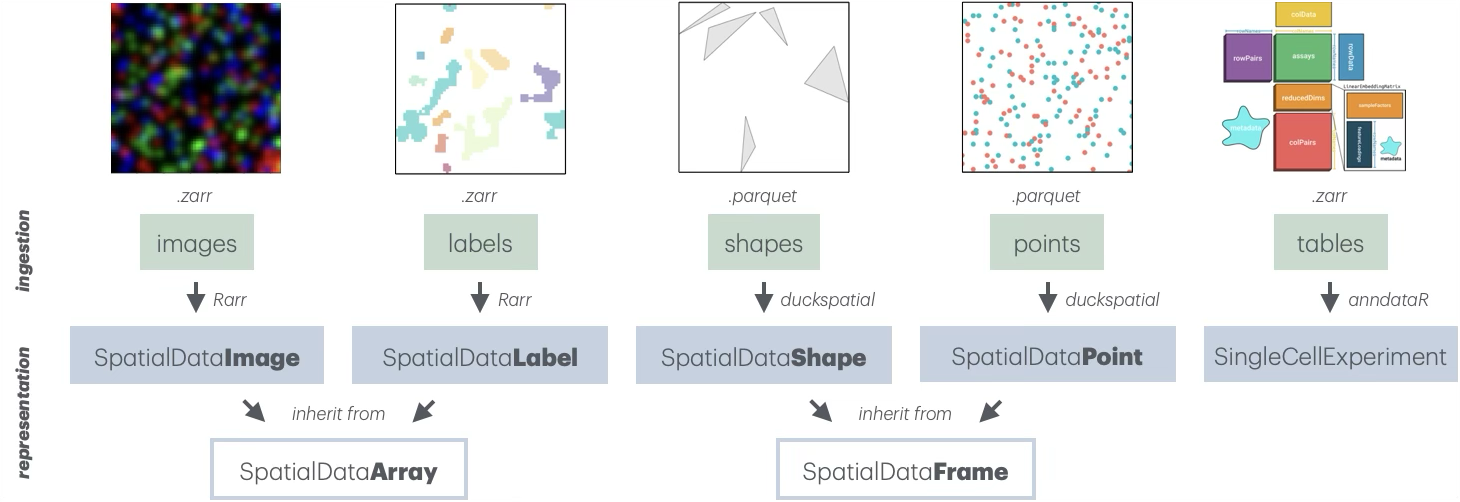
Representation
The core data structure is the SpatialData class, which
organizes data into 5 coordinated layers: images, labels,
points, shapes, and tables. Each layer is stored as a list of
layer-specific objects that carry associated
SpatialDataAttr (@meta slot), which encode
spatialdata-specific zarr attributes (.zattr for
Zarr v2, and zarr.json for Zarr v3) Together, these layers
provide a unified representation of spatial omics data, combining
raster, vector, and tabular data within a single coherent framework.
Images/labels store raster-based data as
multi-scale, multi-channel arrays (e.g., immunofluorescent images or
segmentation masks). They are represented as
SpatialDataImage/Label objects that, in turn, inherit from
SpatialDataArray. These are backed by a list of
ZarrArrays via Rarr and
ZarrArray,
enabling chunked, on-disk access.
Points/shapes represent spatial coordinates and
geometric regions (e.g., transcript locations or segmentation
boundaries). They are represented as SpatialDataPoint/Shape
objects that, in turn, inherit from SpatialDataFrame. These
are DuckDB-backed by a duckspatial_df, enabling efficient
lazy handling.
Tables store functional annotations or information that has been aggregated across layers (e.g., gene cell data). They are currently represented as in-memory SingleCellExperiment objects; delayed, Zarr-backed handling of assay data is under active development.
Handling
SpatialData are represented on-disk as Zarr stores. The
package provides the readSpatialData() function to ingest
an entire store, although arguments to control which layers and elements
to read or not to read are also available.
For this demonstration, we use a toy dataset included in the package:
# dependencies
library(SpatialData)
library(SingleCellExperiment)
# path to 'spatialdata' Zarr store
zs <- file.path("extdata", "blobs.zarr")
zs <- system.file(zs, package="SpatialData")
# read as 'SpatialData' R object
(sd <- readSpatialData(zs))## class: SpatialData
## - images(2):
## - blobs_image (3,64,64)
## - blobs_multiscale_image (3,64,64)
## - labels(2):
## - blobs_labels (64,64)
## - blobs_multiscale_labels (64,64)
## - points(1):
## - blobs_points (200)
## - shapes(3):
## - blobs_circles (5,circle)
## - blobs_multipolygons (2,polygon)
## - blobs_polygons (5,polygon)
## - tables(1):
## - table (3,10) [blobs_labels]
## coordinate systems(5):
## - global(8): blobs_image blobs_multiscale_image ... blobs_polygons
## blobs_points
## - scale(1): blobs_labels
## - translation(1): blobs_labels
## - affine(1): blobs_labels
## - sequence(1): blobs_labelsThe output above summarizes the SpatialData object,
showing the dimension of elements in each layer (images, labels, points,
shapes, tables), as well as the defined coordinate systems and which
elements align to each of these.
Accession
SpatialData objects behave like a nested list: the first
level corresponds to layers, and the second level corresponds to
elements within each layers. For convenience, frequently needed accessor
functions are provided as well.
We here demonstrate various equivalent ways of accessing a layer or element:
# preferred way
# (using accessor)
image(sd, 1)
image(sd, "blobs_image")
# alternative ways
# (using list-style)
images(sd)[[1]]
images(sd)[["blobs_image"]]
sd$images[[1]]
sd$images$blobs_image
sd$images[["blobs_image"]]Similarly, the following are equivalent ways of retrieving element names:
# preferred way
# (using accessor)
imageNames(sd)
# alternative ways
# (using list-style)
names(sd[[1]])
names(sd$images)
names(sd[["images"]])Subsetting
Object-wide subsetting of SpatialData is supported via
[, which can be used to drop layers, or elements within
layers. Note that the following operations are not in-place, but return
a SpatialData object with fewer layers/elements:
Internals
Every spatial element (tables excluded) is composed of two key slots:
data: a list ofZarrArrays for images/labels, or aduckspatial_dffor shapes/points.meta: aSpatialDataAttrsobject containing the OME-NGFF metadata retrieved from the zarr attributes present in the original Zarr store.
We here demonstrate how to access these slots for a given element
image elements represent a special case, as they are
stored as a list of ZarrArrays (one per multi-scale
resolution). For them, data() provides an additional
argument k that specifies which resolution to retrieve:
-
k=1retrieves the highest resolution (default). -
k=Infretrieves the lowest resolution available. -
k=NULLretrieves the full list of available resolutions.
# get multi-scale image
i <- image(sd, 2)
a <- data(i, 1) # highest
b <- data(i, Inf) # lowest
# available resolutions
l <- data(i, NULL)
# dimensions of each
d <- vapply(l, dim, integer(3))
rownames(d) <- c("c", "y", "x")
colnames(d) <- seq_along(l)
show(d)## 1 2 3
## c 3 3 3
## y 64 32 16
## x 64 32 16Annotations
For single-cell and spatial omics datasets, functional annotations
are commonly stored as AnnData objects in Python. In
R, we use anndataR
(Deconinck et al.
2025) to read these Zarr-backed AnnData as SingleCellExperiment(s).
A table can link to one or more label or
shape (but not other layers), whereby internal metadata
(spatialdata_attrs) are used to keep track of the
element(s) and observations being annotated. This is handled internally
so the user needn’t worry about it; however, we show it here for
didactic purposes:
# access annotation
(se <- table(sd))## class: SingleCellExperiment
## dim: 3 10
## metadata(0):
## assays(1): X
## rownames(3): channel_0_sum channel_1_sum channel_2_sum
## rowData names(0):
## colnames(10): 3 4 ... 15 16
## colData names(0):
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
# annotated region(s)
region(se)## [1] "blobs_labels"
# annotated instance(s)
instances(se)## [1] 3 4 5 8 10 11 12 13 15 16Transformations
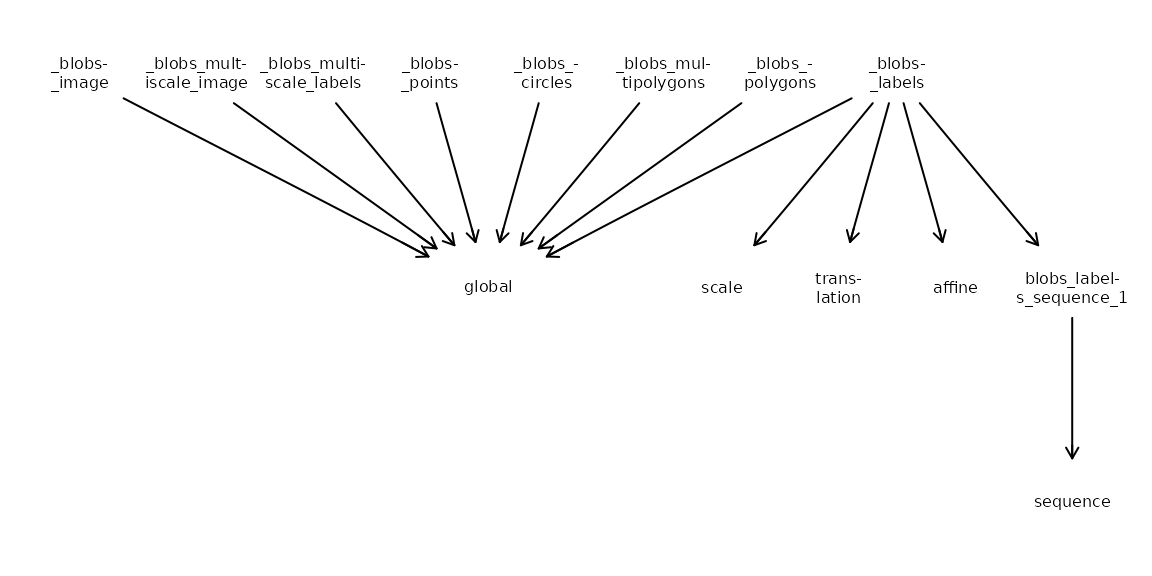
A key feature of the SpatialData framework is its
handling of different coordinate systems. Each element can exist in
multiple coordinate spaces simultaneously, defined by transformations in
its on-disk Zarr attributes.
The relationships between different elements and their respective
coordinate spaces can be complex. SpatialData provides the
CTgraph() and CTplot() functions to construct
and visualize a directed graph of these relationships:
-
source nodes (prefixed with
_) represent individual elements. -
target nodes represent the coordinate systems
(e.g.,
global). - edges represent the transformations required to align an element.
The transform() function resolves the necessary steps to
project an element into a target coordinate system by traversing this
graph, and applying the respective transformation(s). Under the hood,
this involves:
Retrieving the relevant transformation data from the element’s
SpatialDataAttr(e.g., scale factors for x- and y-coordinates); and,Applying the appropriate transformation function(s) in the correct order (e.g.,
scale()thentranslation()).
# get element
a <- label(sd)
# project into 'global'
b <- transform(a, "scale")
# compare XY extents
do.call(rbind, c(a=extent(a), b=extent(b)))## [,1] [,2]
## a.x 0 64
## a.y 0 64
## b.x 0 128
## b.y 0 192Utilities
Cropping
crop() may be used to subset elements – across all
layers – according to a spatial bounding box or polygon. This
region may be supplied in different ways, including as a
SpatialDataShape. In addition, the following are okay:
For bounding box cropping, an
sf::st_bbox()object, or a list ofxmin/xmax/ymin/ymaxvalues (order irrelevant).For polygon cropping, an
sf::st_polygon()orsf::st_sfc()object, or a two-column matrix of XY coordinates (at least 3 rows = triangle).
# bounding box
xy <- list(xmin=-Inf, xmax=Inf, ymin=20, ymax=40)
sp <- crop(sd, xy)
plot(
point(sd)$geometry, col="blue",
main="crop() with\nbounding box")
points(point(sp)$geometry, col="red")
# polygon
xy <- rbind(c(0, 0), c(64, 0), c(32, 64))
sq <- crop(sd, xy)
plot(
point(sd)$geometry, col="blue",
main="crop() with\npolygon")
points(point(sq)$geometry, col="red") 
Masking
mask() aggregates data between elements and across
layers, with support for masking of points by images by labels, points
by shapes, and shapes by shapes:
point by shape masking counts the number of points that fall within each shape (e.g., counting transcripts in cell membrane or nucleus segmentation boundaries in order to obtain a gene cell-level data).
image by label masking aggregates channel-wise pixel values in an image according to the regions defined by a label (e.g., obtaining mean fluorescence intensities per cell).
shape by shape masking aggregates the data in a table of one shape by another shape (e.g., summarizing cell-level data into regions of interest).
A couple considerations are also worth mentioning:
The identifier of the resulting
tablemay be specified vianame, which will default toi_by_jwhen masking elementiby elementj.Instances of
ithat do not map to any instances ofj(e.g., unassigned transcripts) will be assigned to a special “0” column in the resulting table.
# average channel-wise pixel values by labels
sp <- mask(sd, i="blobs_image", j="blobs_labels")
se <- table(sp, "blobs_image_by_blobs_labels")
assay(se)## 3 x 10 sparse Matrix of class "dgCMatrix"##
## 0 0.1325593 0.11240838 0.05495268 0.13883753 0.1759411 0.05216844 0.1574298
## 1 0.1773876 0.08582868 0.07500563 0.14309153 0.1848081 0.18733438 0.1321635
## 2 0.1171959 0.13570302 0.18691482 0.06592828 0.1295153 0.03637876 0.1985580
##
## 0 0.0225221691 0.134730899 0.10671244
## 1 0.2252634943 0.071093576 0.07564495
## 2 0.0002233054 0.006969089 0.09928612
# count different point species in polygons
sp <- mask(sd, i="blobs_points", j="blobs_polygons")
se <- table(sp, "blobs_points_by_blobs_polygons")
assay(se)## 2 x 6 sparse Matrix of class "dgCMatrix"
## 0 1 2 3 4 5
## gene_a 87 . . 1 2 .
## gene_b 105 2 . 1 . 2
# average shape-level data by other shapes
sp <- mask(sd, i="blobs_polygons", j="blobs_circles")Querying
query() filters elements across all layers based on
table metadata in dplyr-style syntax, where
queries may be passed via the ellipsis (...):
TODO
Combining
combine() can be used to merge two
SpatialData objects into one (or many, via
do.call(list(...), combine)). Here, elements names will be
made unique across objects via make.names(), appending a
suffix to the element names of subsequent objects. Alternatively, names
could be customize before combining.
## original combined
## images 2 4
## labels 2 4
## points 1 2
## shapes 3 6
## tables 1 2
imageNames(sp)## [1] "blobs_image" "blobs_multiscale_image"
## [3] "blobs_image.1" "blobs_multiscale_image.1"Coordinates
centroids() may be used to extract spatial coordinates
for every instance in a given element. This applies all layers except
images and tables. Notably, for labels and shapes, the centroids of each
region are returned (center of mass).
## x y genes
## 1 6 48 gene_b
## 2 41 28 gene_b
## 3 27 54 gene_b
## 4 6 44 gene_a
## 5 13 6 gene_b
## 6 33 61 gene_bextent() will obtain the range of an element’s spatial
coordinates in a target coordinate space. This can be done for one
element, or object-wide in order to obtain the largest extent across all
elements in an object.
## x1 x2 y1 y2
## 0 64 0 64## x1 x2 y1 y2
## 1 62 1 62
# with prior alignment to target coordinate space
xy <- extent(label(sd))
yx <- extent(label(sd), "scale")
rbind(native=unlist(xy), scaled=unlist(yx))## x1 x2 y1 y2
## native 0 64 0 64
## scaled 0 128 0 192Appendix
Resources
SpatialData.plot: companion package with
ggplot-based visualization capabilities, including layered plotting of different elements with control over each’s aesthetics, channel-mixing and auto-contrasting for images, transformations handling, etc.SpatialData.data: companion package with example datasets from different platforms, including use of BiocFileCache for efficient data management.
SpatialData.demo: companion repository with vignettes on analyzing datasets from different platforms, including transcriptomics and proteomics.
Session info
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] SingleCellExperiment_1.34.0 SummarizedExperiment_1.42.0
## [3] Biobase_2.72.0 GenomicRanges_1.64.0
## [5] Seqinfo_1.2.0 IRanges_2.46.0
## [7] S4Vectors_0.50.1 BiocGenerics_0.58.1
## [9] generics_0.1.4 MatrixGenerics_1.24.0
## [11] matrixStats_1.5.0 SpatialData_0.99.37
## [13] BiocStyle_2.40.0
##
## loaded via a namespace (and not attached):
## [1] tidyselect_1.2.1 EBImage_4.54.0 blob_1.3.0
## [4] dplyr_1.2.1 R.utils_2.13.0 bitops_1.0-9
## [7] fastmap_1.2.0 RCurl_1.98-1.18 duckdb_1.5.2
## [10] digest_0.6.39 lifecycle_1.0.5 sf_1.1-1
## [13] paws.storage_0.9.0 magrittr_2.0.5 compiler_4.6.0
## [16] rlang_1.2.0 sass_0.4.10 tools_4.6.0
## [19] yaml_2.3.12 knitr_1.51 S4Arrays_1.12.0
## [22] htmlwidgets_1.6.4 classInt_0.4-11 curl_7.1.0
## [25] reticulate_1.46.0 DelayedArray_0.38.1 KernSmooth_2.23-26
## [28] abind_1.4-8 withr_3.0.2 purrr_1.2.2
## [31] desc_1.4.3 R.oo_1.27.1 grid_4.6.0
## [34] e1071_1.7-17 cli_3.6.6 rmarkdown_2.31
## [37] crayon_1.5.3 ragg_1.5.2 proxy_0.4-29
## [40] DBI_1.3.0 cachem_1.1.0 BiocManager_1.30.27
## [43] XVector_0.52.0 tiff_0.1-12 geoarrow_0.4.2
## [46] vctrs_0.7.3 Matrix_1.7-5 jsonlite_2.0.0
## [49] bookdown_0.46 fftwtools_0.9-11 RBGL_1.88.0
## [52] Rgraphviz_2.56.0 systemfonts_1.3.2 jpeg_0.1-11
## [55] locfit_1.5-9.12 jquerylib_0.1.4 units_1.0-1
## [58] glue_1.8.1 pkgdown_2.2.0 ZarrArray_1.0.0
## [61] Rarr_2.1.8 tibble_3.3.1 pillar_1.11.1
## [64] rappdirs_0.3.4 nanoarrow_0.8.0 htmltools_0.5.9
## [67] graph_1.90.0 dbplyr_2.5.2 R6_2.6.1
## [70] httr2_1.2.2 wk_0.9.5 textshaping_1.0.5
## [73] evaluate_1.0.5 lattice_0.22-9 R.methodsS3_1.8.2
## [76] png_0.1-9 duckspatial_1.0.0 paws.common_0.8.9
## [79] bslib_0.11.0 class_7.3-23 uuid_1.2-2
## [82] Rcpp_1.1.1-1.1 SparseArray_1.12.2 anndataR_1.2.0
## [85] xfun_0.57 fs_2.1.0 pkgconfig_2.0.3